Le format VCF
En résumé
- Un VCF est une liste de variations
- Ses colonnes de base sont CHROM POS ID REF ALT INFO
- Une information manquante est généralement matérialisée par un
. - FILTER précise si la variation passe les critères de qualité fixés par le bioinformaticien lors du calling
- INFO est la colonne contenant les annotations pour une variation
- FORMAT précise l'ordre des informations des échantillons
Le format VCF pour Variant Call Format est un fichier texte tabulé listant les variations d'une séquence comparé à une référence.\ Dans le cadre de Diagho, les variations listées seront celles observées dans l'ADN d'un patient et de ses apparentés et le VCF servira donc de fichier d'entrée principal.
Un fichier VCF ne contient cependant pas nécessairement de données de séquençage et peut tout autant servir de source pour des annotations comme le fait ClinVar par exemple.
À l'inverse, un VCF peut très bien contenir les données de séquençage de centaines d'échantillons et aucune annotation particulière.
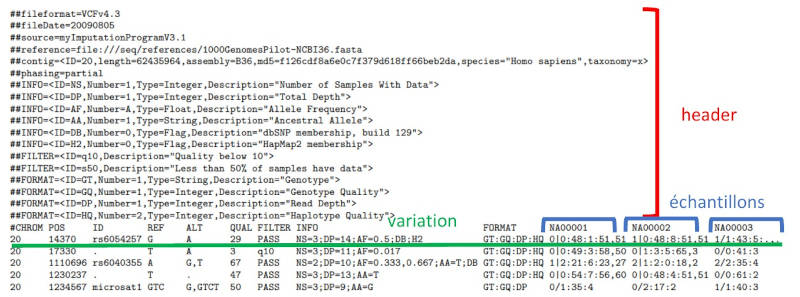
Le header
Il est constitué des lignes débutant par ## et peut contenir tout type d'informations.
Typiquement elles servent à préciser les critères des FILTER, la définition des FORMAT
et le contenu des INFO.\
On retrouve également souvent la liste des contigs du génome de référence parcourus lors
du variant calling.
Attention
Seules deux lignes sont obligatoires dans le header :
##fileformat [...] et #CHROM POS [...].\
Bien que contre-productif, rien n'empêche un VCF de ne pas avoir de header autre que ces
deux lignes.
Les champs INFO
Les champs INFO permettent de préciser le contenu et le format des annotations ajoutées dans la colonne INFO du VCF. Étant un header optionnel, il est peu prudent de n'espérer dépendre que d'eux, mais lorsqu'ils sont renseignés, ils suivent normalement un formalisme assez précis.
##INFO=<ID=ID,Number=number,Type=type,Description="description",Source="source",Version="version">
- ID sera la valeur affichée comme nom d'annotation
- Number est un entier indiquant le nombre d'annotations par variation\ Plusieurs cas particuliers :
- A : une valeur par ALT
- R : une valeur par ALT + REF
- G : une valeur par GT
- 0 : pas de valeur, seule l'ID est ajouté (pour un flag typiquement)
- . : nombre de valeurs variable ou inconnu
- Type : peut être Integer, Float, Flag, Character ou String
- Description : doit être entourée de double-quote
" - Source (rare) : origine de l'annotation
- Version (rare) : version de la source de l'information
Les colonnes
CHROM POS REF ALT
Le chromosome, la position, la base sur le génome de référence à cette position et
l'alternative observée lors du variant calling.\
Plusieurs alternatives peuvent être appelées sur une même position. Elles sont alors séparées
par une , si le fichier n'a pas été décomposé.
ID
Un identifiant de la variation ou de la position. Il n'y a pas de protocole strict à
respecter mais si identifiant il y a, c'est presque systématiquement un Reference SNP de
la dbSNP (sous la forme /^rs\d+$/).
En cas d'absence d'ID, il est remplacé par un .
QUAL
C'est un score de qualité correspondant à un score Phred estimant la probabilité que la
position ne porte en réalité pas de variation.\
QUAL = -10log(10)
- si QUAL = 20, il y a 99% de chance que la position porte bien une variation
- si QUAL = 30, il y a 99,9% de chance que la position porte bien une variation
- si QUAL = 3, il y a 50% de chance que la position porte bien une variation
FILTER
Ce champ permet de préciser si la variation relevée passe les critères de qualité exigés
par le bioinformaticien lors du calling et spécifiés dans le header (s'il y en a). Par
exemple, dans le screenshot ci-dessus, la seconde variation a une QUAL inférieure à 10.\
S'il existe plusieurs FILTER que la variation ne respecte pas, ils seront tous notés dans
la colonne et séparés par des ,
INFO
Cette colonne contient toutes les annotations d'une variation, chacune étant séparée par
un ;. Les annotateurs produisant des VCF ajoutent leurs
informations dans cette colonne.
Ces infos ne sont pas nécessairement issues de base de données mais peuvent également
provenir du séquençage lui-même, de l'alignement, du calling, des échantillons… Par
exemple, l'AF (Allele Frequency) précise le ratio ALT / (REF + ALTs) des
échantillons du VCF.
Cette colonne peut également servir de source d'annotations indépendamment de la présence d'échantillon ou non.
FORMAT
Lorsque le VCF contient des échantillons, cette colonne précise la structure des informations des colonnes des échantillons, le détail de ces informations étant normalement précisé dans le header.\ Ainsi, d'après l'exemple ci-dessus, sur la première variation :
0|0est leGTdeNA0000148est sonGQ1est saDP51,51est saHQ
Notez avec la 4e variation que le FORMAT peut varier au sein du VCF.
Échantillons ou samples
Chaque colonne au delà de FORMAT correspond aux informations de séquençage d'un échantillon, typiquement le génotype d'un patient (présence ou non de la variation chez des individus). Le nom de la colonne correspond à l'ID d l'échantillon.
L'information la plus importante de ces colonnes dans notre contexte est le génotype (GT) :
- 0/0 signifie que l'échantillon a une REF sur ses deux chromosomes
- 0/1 signifie que l'échantillon a une REF sur un chromosome et porte l'ALT sur le second
- 1/1 signifie que l'échantillon porte l'ALT sur ses deux chromosomes
Dans le cas d'un VCF non décomposé, 1/2 signifie qu'il porte la première ALT sur un chromosome et la seconde ALT sur le second chromosome.
[^1]: Les spécifications du format VCF