Interprétation
Ceci n'est qu'un exemple naïf du déroulement d'une interprétation de données génomiques pour en présenter sommairement les grandes étapes.
Note
La compréhension de cette page sera peut-être facilitée par la lecture préalable de l'article concernant les bases du fonctionnement de l'ADN
Les deux chapitres les plus importants sont l'application de filtres et la fouille manuelle des variations.
Étapes préliminaires
Lorsqu'un médecin estime qu'un de ses patients devrait bénéficier d'un diagnostic
génétique, il redirige ce dernier vers un généticien clinicien qui va faire son tableau
clinique (lister tous ses traits phénotypiques et symptomes).
Si le besoin d'un séquençage est confirmé, on effectue un prélèvement sanguin au patient
et à ses parents si possible, prélèvements qui seront ensuite préparés pour un
séquençage.
Une fois le séquençage effectué, le fichier brut de sortie est préalablement traité afin
d'obtenir un fichier 1 qui pourra être annoté et servira de fichier d'entrée sur
l'application Diagho. C'est à partir de ce VCF annoté que peut être menée l'étape
d'interprétation du biologiste en génétique.
Dossiers patients
La première étape est bien entendu de connaître ce dont souffre le patient, ce qui est
permis par les observations du clinicien, photographies du patient, et éventuels
résultats d'analyses, d'imageries…
Chacune de ces informations peut aider à aiguiller le diagnostic, certains syndromes
ayant un tableau clinique si caractéristique qu'ils ne nécessiteraient même pas de
séquençage et permettant au généticien de savoir exactement où chercher, tandis que des
tableaux très flous comme « DI non syndromique » promettent au biologiste une longue
journée.
Ces informations restent pertinentes tout le long de l'interprétation. Par exemple, en
lisant une publication sur une variation, le généticien peut vouloir facilement
réafficher la photo du patient pour le comparer avec d'autres patients porteurs de la
variation considérée.
Leur offrir la possibilité de facilement réaccéder à ce dossier avant comme pendant
l'interprétation a donc tout son sens.
Contrôles qualité ou QC
Cette étape n'est pas nécessairement menée par le généticien même, mais elle permet de s'assurer que le fichier obtenu pour mener l'interprétation n'a pas subi d'altérations rendant son exploitation impossible (que ce une erreur de manipulation dans la préparation des échantillons, un soucis au niveau du séquençage ou un possible bug informatique).
Application de filtres
C'est un des cœurs de l'application. La liste de variations de base est enrichie
d'informations complémentaires comme le gène où se situe la variation ou à quel point
elle est courante dans la population.
Ces informations aident à faire un tri dans la liste initiale de variations.
???+ example "Exemple" Une variation courante dans la population ne peut par définition
pas être cause d'une maladie rare donc autant les masquer.
À l'inverse, une variation dans un gène connu pour causer une maladie similaire à celle
du patient sera digne d'être étudiée.
Ces filtres sont à adapter en fonction du contexte. Ils peuvent permettre d'appliquer un panel de gènes (afficher uniquement les variations présentes uniquement dans une liste précise de gènes) ou au contraire rester très généraliste.
Ces filtres sont ensemblistes et doivent pouvoir faire leur tri sur différents critères simultanément, le tout organisé par connexions logiques.
???+ example "Exemple" Afficher les variations :
connues comme étant pathogènes
OU présentes dans le panel de gènes "XYZ"
OU (présentes chez moins de 0.1% de la population ET avec un score de prédiction
délétère)
Enfin ces filtres peuvent être organisés de manière séquencielle telle une succession de tamis qui permettent à l'arrivée de réduire l'ensemble des variations de départ en de courtes listes d'intérêt exploitables par un humain.
Exemples de stratégies de filtrage

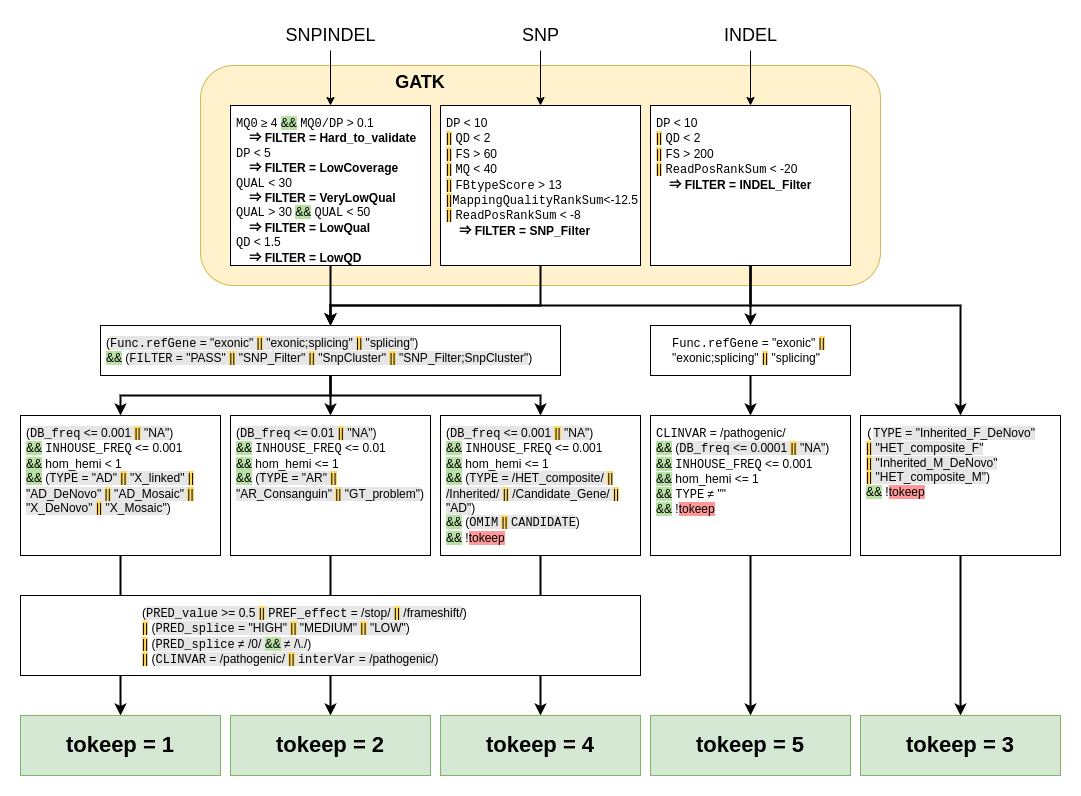
Pour simplifier le travail des généticiens, dans un projet donné, il sera possible de
pré-calculer un ensemble de filtres de départ pour que tout soit directement prêt au
lancement de l'interprétation.
Le généticien doit cependant pouvoir être capable d'appliquer ses propres filtres à la
demande en cours d'interprétation. Cela entraîne deux problèmatiques importantes :
réussir à offrir une UX agréable pour une tâche qui l'est beaucoup moins et pouvoir
adapter les filtres applicables sur des annotations qui varient d'une analyse à l'autre
et sur tous les types de données (strings, nombres absolus, floats entre 0 et 1 ou non,
booléens).
Enfin, outre ces filtres complexes, pour le confort de l'utilisateur, il faudra prévoir une liste prédéfinie de filtres rapides applicables rapidement pour un meilleur confort.
???+ example "Exemple" Alors que le généticien a chargé les variations "récessives liées à l'X" que lui aura réuni une succession de filtres complexes, il pourra en un simple clic faire un tri rapide sur ce tableau pour n'afficher que les variations exoniques.
Fouille des variations
Une fois le plus gros du tri effectué automatiquement, le travail du généticien consiste
à effectuer une fouille manuelle des variations restantes pour tenter d'y trouver la
cause moléculaire (la variation/les quelques unes responsables de la maladie).
C'est une étape laborieuse nécessitant le recours à différents outils et sources
d'informations, donc une étape d'autant plus importante pour apporter de la valeur à
l'utilisateur.
Tri manuel
Une première façon de les aider serait de permettre de facilement classer les variations rencontrées au fil de la fouille comme étant non pertinentes (et donc les masquer du tableau) ou au contraire de les garder pour une enquête ultérieure plus approfondie.
??? abstract "Use case" Pour prendre un fonctionnement sur Excel, le généticien peut sélectionner la première cellule de la colonne "OMIM" (les phénotypes associés au gène touché par la variation), garder les yeux rivés sur le contenu de la cellule et rapidement descendre le long de la colonne tant que les phénotypes ne concordent pas avec ceux du patient. Une fois arrivé à un phénotype possiblement concordant avec celui du patient, le généticien peut vouloir garder cette variation pour une enquête approfondie immédiate ou ultérieure.
1 | |
Bases de données externes
L'enquête approfondie évoquée plus haut passe par le recoupage d'informations de sources diverses notamment par le recours à des bases de données externes amassant de gros volumes de données sur les gènes, les maladies génétiques et même les variations rencontrées par les laboratoires du monde entier, comme leurs nombres d'occurrence dans une population ou le fait qu'elles aient déjà été considérées causales d'une maladie.
Exemples de bases de données externes








IGV et les genome browsers
Il est également parfois nécessaire d'observer concrètement la variation dans le
contexte de son séquençage pour comprendre son impact ou vérifier que ce n'est pas un
artefact.
On utilise pour cela des outils appelés genome browsers qui, à partir des fichiers
BAM de l'alignement1, offrent entre autres une représentation graphique de tous les
reads alignés sur le génome de référence.
Le plus connu et utilisé est IGV et existe sous forme de client et de lib JS.
Genome browser


Revue de la littérature
Enfin, une dernière étape est la vérification de l'existance de publications traitant de
la variation visée, typiquement via Pubmed. Si
d'autres chercheurs à travers le monde sont arrivés aux mêmes conclusions vis à vis
d'une variation précise, cela devient un argument de poids pour le rendu du
diagnostic.
Il peut être intéressant de pouvoir inclure facilement le DOI de ces publications dans
les étapes suivantes de reporting.
Classification des variations
Les laboratoires du monde entier ont intégré une nomenclature précise quant à l'évaluation de la pathogénicité des variations rencontrées. Elle suit les recommandations de l'ACMG qui répartissent les variations en 5 classes de pathogénicité croissante :
- Benign
- Likely benign
- Uncertain significance
- Likely pathogenic
- Pathogenic
Le concept de "likely" (ou "probablement") sous-tend une créance supérieure à 90%. Cette
classification doit être incluse sur le compte-rendu d'interprétation à destination du
prescripteur.
Cette classification suit des règles précises pour être aussi objective que possible ;
plus de détails dans l'article dédié à l'ACMG.
Génération de compte-rendu
La finalité de l'interprétation est de générer un compte-rendu d'analyse à renvoyer au prescripteur pour rendre compte des variations pathogènes trouvées ou non. Ils doivent se conformer à un format assez précis, et bien que Diagho puisse simplifier et automatiser son remplissage, certains laboratoires préfèreront utiliser leur SGL en place pour cette tâche.
Un autre fichier de sortie possible est le rapport d'interprétation. Bien moins
formaté, son but est de rendre compte des observations du généticien à ses collègues
lors d'une RCP.
Il est donc possible de faire gagner un temps précieux aux utilisateurs à cette étape en
mettant en place une pré-complétion de templates .pptx ou .docx en facilitant l'ajout
des variations retenues et toutes leurs nomenclatures associées2, de commentaires
personnels, de screenshots (genome browser, BDD ou graph), de liens vers des
publications…